|
|
|
|
On both symmetric multiprocessors (SMPs) and massively parallel processors (MPPs), MercuryDPM can run simulations in parallel. The distinction between the computer architectures is that SMPs (single-processor machines) use shared memory, while MPPs (clusters) use distributed memory. We parallelise on shared memory with an OpenMP implementation and parallelize on distributed memory with an MPI implementation. The following tutorials will demonstrate how to use MPI parallelisation in your code.
Before we get into the implementation, it's important to note that the MercuryDPM-MPI algorithm employs a simple domain decomposition technique. The decomposition splits the domain into \(n_x \times n_y \times n_z\) subdomains and running each subdomain on a separate processor.
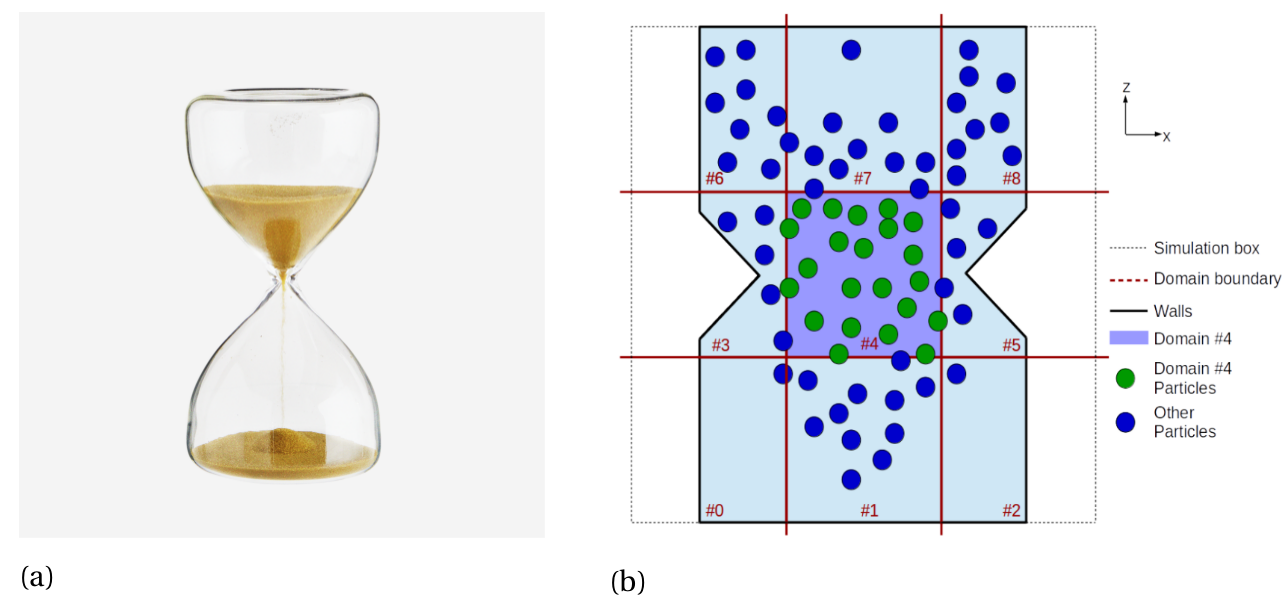
In the figure below, we show an actual hourglass filled with sand, and a corresponding sketch of a (two-dimensional) hourglass simulation, depicting a spatial decomposition using a Cartesian grid of \(3 \times 1 \times 3\), using a total of 9 threads. The decomposition splits the domain (defined via setMin and setMax in MercuryDPM) into intervals of equal size in each Cartesian direction. It is worth noting that the subdomains on the boundary of the domain extend towards infinity, thus even particles outside the domain belong to one of the subdomains. MPI is a distributed memory-approach, meaning all particles located inside a domain are only known to the thread simulating that particular subdomain. Note, we only distribute the particles, not the walls and boundaries, since they consume considerably less storage than the particles. For more information, see [1].
To run simulations in parallel, you need to define all particles in setupInitialConditions and you need to define the domain size in main(). Assume your driver code is structured as follows:
To run your simulation in parallel, you need to compile the code with MPI. Use cmake and turn the flag MercuryDPM_USE_MPI to ON. You can do this either by loading cmake-gui and changing the use USE_MPI flag to ON; or, alternatively you do it on the command line:
You also need to tell your program about the decomposition it should use. To split your domain setNumberOfDomains into \(n_x \times n_y \times n_z\), add the following command in your main function before solve():
Now compile your code, and run it with mpirun. Make sure you use the correct number of processors ( \(n=n_x \cdot n_y \cdot n_z\)) needed for the domain decomposition:
For an example of an MPI-ready code, see /Drivers/MercurySimpleDemos/HourGlass2DDemoMPI.cpp and Drivers/ParallelDrum/testDrum.cpp.

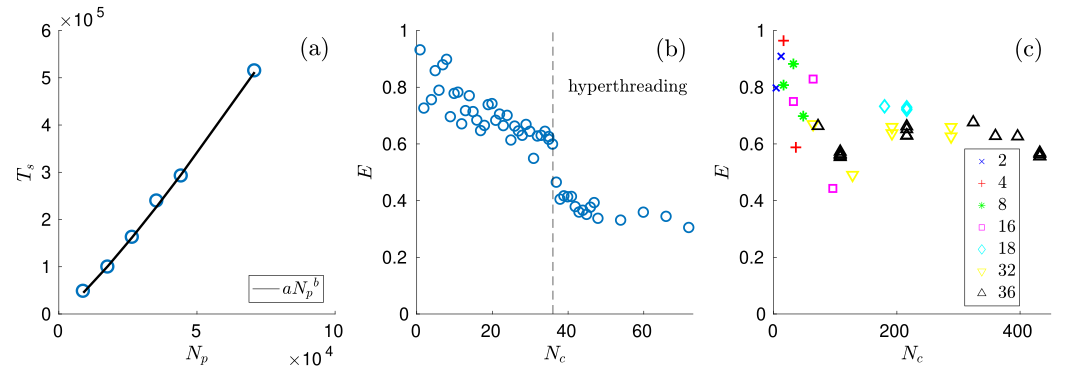
The performance of the MPI parallel algorithm has been tested for the case of a rotating drum of varying width; a snapshot of the simulations is shown in the above Figure. The serial algorithm shows a near-linear scaling of computing time with the number of particles (Figure (a) shown below). Weak scaling of the parallel implementation is shown by measuring the efficiency E = Ts/(Tp*Nc), the ratio of computing time for the serial and parallel implementation, for a varying number of cores Nc, keeping the number of particles per core constant. On a single node, efficiency decreases slowly with the number of cores, but levels off at around 40% for simulations that use hyperthreading (Figure (b) shown below). On multiple nodes, hyper-threading can be avoided, and the efficiency remains above 60% (Figure (c) shown below). Thus, the algorithm performs efficiently for large simulations, if the computational load per core is homogeneous.
On the following pages, you'll find more parallelisation demonstrations:
Parallel processing using OpenMP
Parallel processing for Input-Output, I/O files
Alternatively, go back to Overview of advanced tutorials